Genetische achtergrond van Sikkelcelziekte
Sikkelcelziekte is een erfelijke ziekte. Het erfelijk materiaal (het DNA) bevindt zich op de chromosomen. Ieder kind krijgt 46 chromosomen van beide ouders; 22 chromosomen en één geslachtschromosoom van de moeder en 22 chromosomen en één geslachtschromosoom van de vader. De chromosomen zijn de dragers van de genen. Door een verandering in een gen (mutatie) op de korte arm van chromosoom 11 in het gen voor bèta globine ontstaat bij sikkelcelziekte een verandering in het erfelijk materiaal.

Genen die te maken hebben met de productie van HbA1 en HbF liggen dichtbij elkaar op chromosoom 11. Hemoglobine is gemaakt van twee van de vier genen uit het alfa globine gen cluster op chromosoom 16 en twee genen uit het bèta globine gen cluster op chromosoom 11. Vier globine ketens: twee alpha globine ketens en twee bèta globine ketens vormen samen het HbA1 hemoglobine, het belangrijkste type hemoglobine vanaf de leeftijd van vier maanden.
De compound heterozygote sikkelcelziekte aandoeningen ontstaan wanneer je in beide bèta globine genen een verschillende verandering hebt i.t.t. bij de homozygote sikkelcelziekte waarbij je in beide bèta globine genen dezelfde S verandering hebt (HbSS).
Deze compound heterozygote of “samengestelde” sikkelcelziekten kunnen in de volgende vormen voorkomen;
- Hemoglobine SC sikkelcelziekte: één ouder geeft het HbS gen door, en de andere ouder het HbC gen; het kind heeft dan HbSC sikkelcelziekte. Hierdoor ontstaan klachten die vergelijkbaar zijn met de klachten passende bij HbSS sikkelcelziekte. Over het algemeen zijn de klachten vaak iets milder van aard. De behandeling verschilt echter niet. De klachten bij deze aandoening zijn wel ernstiger dan bij homozygote HbC (HbCC) ziekte.
- Sikkelcel- bèta-thalassemie ziekte: één ouder geeft het HbS gen door en een ouder het gen voor bèta-thalassemie. Je krijgt dan een combinatie van sikkelcelziekte en bèta-thalassemie oftwel HbS bèta-thalassemie sikkelcelziekte. Je hebt dan ook klachten die bij beide ziekten passen, de ernst van de klachten kan erg wisselen afhankelijk van de hoeveelheid normaal hemoglobine die nog geproduceerd wordt door de bèta globine genen. Als er bijna geen normaal hemoglobine wordt gevormd (HbS bèta 0-thalassemie) is er sprake van ernstig beeld. Als er toch een redelijke hoeveelheid normaal hemoglobine wordt gevormd (HbS bèta + - thalassemie) dan zullen de klachten vaak minder ernstig zijn, met o.a. minder sikkelcelcrisen. Dit type sikkelcelziekte wordt vooral gezien bij mensen die oorspronkelijk afkomstig zijn uit het Mediterrane gebied (Italië, Griekenland, Marokko, Turkije). Soms staat bij dit type sikkelcelziekte de bloedtransfusie behoefte op de voorgrond en verloopt de ziekte zoals bij een ernstige bèta thalassemie (of bèta thalassemia major).
-
Sikkelcelziekte is een erfelijke bloedziekte die veroorzaakt wordt door een afwijkende bouw van het hemoglobine in de rode bloedcel. Hemoglobine is een eiwit dat in de rode bloedcel zit. Het is belangrijk voor het zuurstoftransport in je lichaam.
-
Sikkelcelziekte is een erfelijke ziekte. Het erfelijk materiaal (het DNA) bevindt zich op de chromosomen.
-
Sikkelcelziekte komt overal voor, maar voornamelijk in Afrika en de landen waar veel mensen wonen die oorspronkelijk uit Afrika komen. Vooral uit gebieden waar al van oudsher malaria heerst.
-
De problemen die kunnen ontstaan bij sikkelcelziekte worden niet alleen veroorzaakt door de sikkelcellen, ook de veranderde werking van de bloedvaatwand speelt een rol.
-
Hoeveel klachten je hebt verschilt van persoon tot persoon. Omdat de klachten afhankelijk zijn van verschillende factoren zoals: het type sikkelcelziekte, hoe erg je bloedarmoede is, of de afbraak van rode bloedcellen, hoe vaak je een sikkelcelcrisis hebt of infecties, en hoe ernstig die zijn.
-
Naast verschillende behandelingsmogelijkheden, is een gezonde leefwijze van belang, vooral als je sikkelcelziekte hebt. Als je gezond leeft kan je lichaam een goede conditie opbouwen. Gezonde voeding, veel drinken, op tijd naar bed en sporten helpen hieraan mee.
-
De diagnose sikkelcelziekte kan gesteld worden op verschillende manieren; met de hielprikscreening vlak na de geboorte, door antenataal genetisch onderzoek of door genetisch familieonderzoek of door bloedonderzoek bij de patiënt zelf.
-
Waar moet je aan denken als je op reis gaat?
-
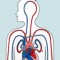
Bloed is het transportsysteem van je lichaam. Het stroomt door je hele lichaam en zorgt voor de aan- en afvoer van stoffen.
-
Om je lichaam te voorzien van zuurstof en voedingsstoffen en om stoffen af te voeren, moet je bloed voortdurend blijven stromen. Het pompen van je hart zorgt daarvoor. We noemen dit de bloedsomloop.
-
Bloed bestaat uit rode bloedcellen, verschillende soorten witte bloedcellen, bloedplaatjes en plasma. Ze hebben allemaal een eigen functie.
-
Bloedcellen hebben een beperkte levensduur. Rode bloedcellen leven 't langst, ongeveer vier maanden. Witte bloedcellen leven gemiddeld enkele dagen en bloedplaatjes zo’n acht tot tien dagen.
-
Ieder mens heeft bloed van een bepaalde bloedgroep. Ook jij.