De diagnose hemofilie
Het moment waarop de diagnose hemofilie wordt gesteld kan verschillen. Als hemofilie in de familie voorkomt, kun je in sommige ziekenhuizen al voor de geboorte onderzoek laten doen. De diagnose kan ook direct na de geboorte worden gesteld of uitgesloten door stollingsonderzoek bij de baby te doen (vaak uit het navelstrengbloed).
Als ze denken aan een stollingsstoornis, word je door de huisarts doorverwezen naar de kinderarts. De kinderarts stelt eerst een aantal vragen, over je bloedingen en over gezondheidsproblemen in je familie. Daarna word je lichamelijk onderzocht en kijken ze naar je blauwe plekken, je slijmvliezen en je spieren en gewrichten. Ook wordt je bloed onderzocht, er wordt gekeken naar de werking van je bloedstolling. Meestal wordt ook nog genetisch onderzoek verricht, om de diagnose te bevestigen.
5.1 Bloedonderzoek
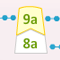
Eerst krijg je een prik om bloed af te nemen. Met het bloed worden verschillende stollingstesten gedaan, zoals een test die de APTT heet. Er wordt altijd naar het bloedbeeld en het aantal bloedplaatjes gekeken, en als ze denken aan hemofilie wordt er ook gekeken hoeveel Factor VIII (8) en IX (9) er in je bloed zit.Je hebt hemofilie A als je minder dan 40% Factor VIII (8) in je bloed hebt, met een normale hoeveelheid VWF, óf als genetisch onderzoek een afwijkende genmutatie voor Factor VIII (8) laat zien.
Je hebt hemofilie B als je minder dan 40% Factor IX (9) in je bloed hebt, óf als genetisch onderzoek een afwijkende genmutatie voor Factor IX (9) laat zien.
Je bent drager van hemofilie als genetisch onderzoek een afwijkende genmutatie voor Factor VIII (8) of Factor IX (9) laat zien.
Bepalingen bij bloedonderzoek
| Bepaling | Wat meet je? | Hemofilie A | Hemofilie B |
| APTT Geactiveerde partiële tromboplastinetijd | Geeft stollingstijd aan, wordt uitgedrukt in seconden. | Verlengd. Bij milde hemofilie slechts zeer gering verlengd. | Verlengd. Bij milde hemofilie slechts zeer gering verlengd. |
| PT Prothrombinetijd | Geeft stollingstijd aan, wordt uitgedrukt in seconden, of als een ratio (INR). | Normaal. | Normaal. |
| Bloedbeeld | Het aantal cellen wordt bekeken; het aantal rode bloedcellen, witte bloedcellen en bloedplaatjes. Ook het volume dat de rode bloedcellen in het bloed innemen wordt bepaald, het hematocriet. | ||
| Bloedplaatjes of trombocyten | Het aantal bloedplaatjes in het bloed wordt gemeten. Wordt uitgedrukt in aantal x 109/l. | Normaal. | Normaal. |
| Factor VIII (8) | De hoeveelheid wordt uitgedrukt in de hoeveelheid (IU) per ml bloed of een percentage dat normaal in bloed aanwezig is. Normaal = 100%. | Verlaagd. | Normaal. |
| Factor IX (9) | De hoeveelheid wordt uitgedrukt in de hoeveelheid (IU) per ml bloed of een percentage dat normaal in bloed aanwezig is. Normaal = 100%. | Normaal | Verlaagd |
| Von Willebrandfactor (VWF) | De hoeveelheid en de werking worden getest. Wordt uitgedrukt in de hoeveelheid (IU) per ml bloed of in percentage. | Normaal. | Normaal. |
Factor VIII (8) kan direct na de geboorte worden gemeten, de hoeveelheid Factor VIII (8) is dan al op zijn hoogste punt waardoor meestal direct duidelijk is of het normaal of te weinig is. Bij sommige milde vormen van hemofilie A is het moeilijker om kort na de geboorte met zekerheid te stellen dat de hoeveelheid verlaagd is, omdat de hoeveelheid Factor VIII (8) stijgt door stress (zoals bij een geboorte).
Factor VIII (8) zit in je bloed vast aan VWF. Zolang Factor VIII (8) aan VWF gebonden is, is de factor niet actief en kan er geen stolling plaats vinden. VWF zorgt er ook voor dat Factor VIII (8) niet door enzymen in je bloed wordt afgebroken. Daarnaast zorgt VWF ervoor dat Factor VIII (8) naar de plek gaat waar bloedstolling nodig is.
Factor VIII (8) is verlaagd bij hemofilie A én bij de Von Willebrandziekte. Bij de Von Willebrandziekte komt dat omdat er onvoldoende VWF is om Factor VIII (8) te beschermen tegen enzymen in je bloed. Als je te weinig Factor VIII (8) hebt, wordt daarom ook altijd de hoeveelheid VWF in je bloed bepaald. Zo wordt duidelijk welke van de twee ziektes je hebt. Soms blijft dit moeilijk en is genetische diagnostiek nodig om erachter te komen of je hemofilie hebt of de Von Willebrandziekte.
De hoeveelheid Factor VIII (8) gaat altijd omhoog bij ziekte, stress of in een andere acute fase. Bij ernstige hemofilie levert dat geen problemen op met het stellen van de diagnose, omdat de hoeveelheid Factor VIII (8) ook tijdens ziekte en stress te laag blijft. Bij milde hemofilie kan dat moeilijker zijn, omdat door ziekte en stress de hoeveelheid Factor VIII (8) tijdelijk is verhoogd
Factor IX (9) kan ook na de geboorte worden bepaald, maar pas na drie maanden is het hoogste punt bereikt. Afhankelijk van de hoogte van Factor IX (9) bij familieleden die ook hemofilie B hebben, kan er een diagnose voor de pasgeborene worden gesteld. Bij milde vormen van hemofilie B is dit moeilijk of nog niet mogelijk. Als het kind bloedingen heeft én Factor IX (9) is sterk verlaagd, dan is dat verdacht voor hemofilie B, maar na drie maanden kun je pas zeker weten of de waarde normaal is of verlaagd.
5.2 Genetisch onderzoek
Met erfelijk onderzoek kun je het defecte gen (de mutatie) vinden dat bij jou hemofilie veroorzaakt. Er zijn verschillende mutaties en het is belangrijk te weten welke jij hebt, vanwege de remmers die je kunt ontwikkelen in de toekomst. Remmers zijn antistoffen die je lichaam zelf aanmaakt tegen Factor VIII (8) of IX (9). Ze zorgen ervoor dat de Factor VIII (8) of IX (9) die je ingespoten krijgt sneller wordt afgebroken. Meestal is het zo dat hoe meer van het gen verdwenen is, hoe minder stollingsfactoreiwit je hebt en hoe groter de kans is op remmers (vooral bij ernstige hemofiliepatiënten). Remmers vormen dus een probleem bij de behandeling van bloedingen met stollingsfactor. Je hebt dan een ander stollingsfactorconcentraat nodig. Ook milde hemofiliepatiënten kunnen remmers ontwikkelen tegen de Factor VIII (8) of IX (9) die ze krijgen. Soms breken de remmers ook het eigen Factor VIII (8) of IX (9) af, waardoor een milde patiënt een ernstige wordt.-
Hemofilie is een aangeboren stollingsziekte waarbij je bloed minder goed stolt. Hierdoor heb je meer last van bloedingen, we noemen dat ook wel een verhoogde bloedingsneiging. Alleen jongens kunnen hemofilie krijgen.
-
Bloedstolling is heel belangrijk, het zorgt ervoor dat een bloeding stopt. Als een bloedvat beschadigd raakt, kan er een gat of scheur in de vaatwand komen, waardoor er bloed naar buiten stroomt. Het bloed komt dan in het weefsel buiten het bloedvat terecht en vormt daar een zwelling en blauwe plek.
-
Bij hemofilie verloopt de eerste fase van de bloedstolling in principe goed. Na een beschadiging hechten de bloedplaatjes zich op normale wijze aan de vaatwand. Het probleem zit in de tweede fase van de bloedstolling.
-
Hemofilie is een zeldzame stollingsziekte die bij jongens voorkomt. Hemofilie A (85%) komt vaker voor dan hemofilie B (15%).
-
Een erfelijke aandoening wordt van ouder op kind doorgegeven, je hebt de ziekte dus al bij je geboorte. Soms krijg je het niet van je ouders, en ben je de eerste in je familie met hemofilie doordat je een nieuwe mutatie (een foutje in je erfelijk materiaal) hebt.
-
Het moment waarop de diagnose hemofilie wordt gesteld kan verschillen. Als hemofilie in de familie voorkomt, kun je in sommige ziekenhuizen al voor de geboorte onderzoek laten doen. De diagnose kan ook direct na de geboorte worden gesteld of uitgesloten door stollingsonderzoek bij de baby te doen (vaak uit het navelstrengbloed).
-
De ene hemofiliepatiënt heeft meer last van bloedingen dan de andere. Dat komt doordat de hoeveelheid Factor VIII (8) of IX (9) die nog in het bloed aanwezig is verschilt van persoon tot persoon.
-
Welke behandeling je nodig hebt, hangt af van de ernst van je bloedingen en de ernst van de verwonding. Het belangrijkste doel van de behandeling is het voorkómen van bloedingen en de schade die je daardoor krijgt, zoals spieratrofie (afbreken van spieren) en gewrichtsschade.
-
Als je hemofilie hebt of een andere stollingsstoornis kun je tegenwoordig, dankzij de medicijnen en de goede zorg, normaal leven en vrijwel alles doen. Er zijn wel paar dingen waar je op moet letten.
-
Al jarenlang wordt veel onderzoek verricht naar hemofilie en mogelijke behandelingen. Eén van de laatste belangrijke ontwikkelingen is gentherapie.